CLINICAL BIOCHEMISTRY SECOND STAGE LECTURE 7 DRTHANA ALSEWEDY INBORN
CLINICALLY RELEVANT ANATOMY 123 ULNAR NERVE ENTRAPMENTLONG ISLAND BHM CONCURRENT CLINICAL PLEASE COMPLETE
PSYCHOLOGY AND CLINICAL LANGUAGE SCIENCES UNIVERSITY OF READING
(INSERT AGENCY NAME) REPRODUCTIVE HEALTH PROGRAM CLINICAL POLICIES AND
006-17%20Clinical%20Psychiatrist%20%20Board%20%20037869
1 COURSE TITLE CLINICAL PRACTICUM IN AUDIOLOGY 2 2
Inborn error in metabolism of amino acids
Clinical
biochemistry second stage lecture 7 Dr.Thana Alsewedy
lecture 7 Dr.Thana Alsewedy
Inborn error in metabolism of amino acids
Inborn errors of amino acid metabolism are metabolic disorders of in which impair the synthesis and degradation .amino acids .
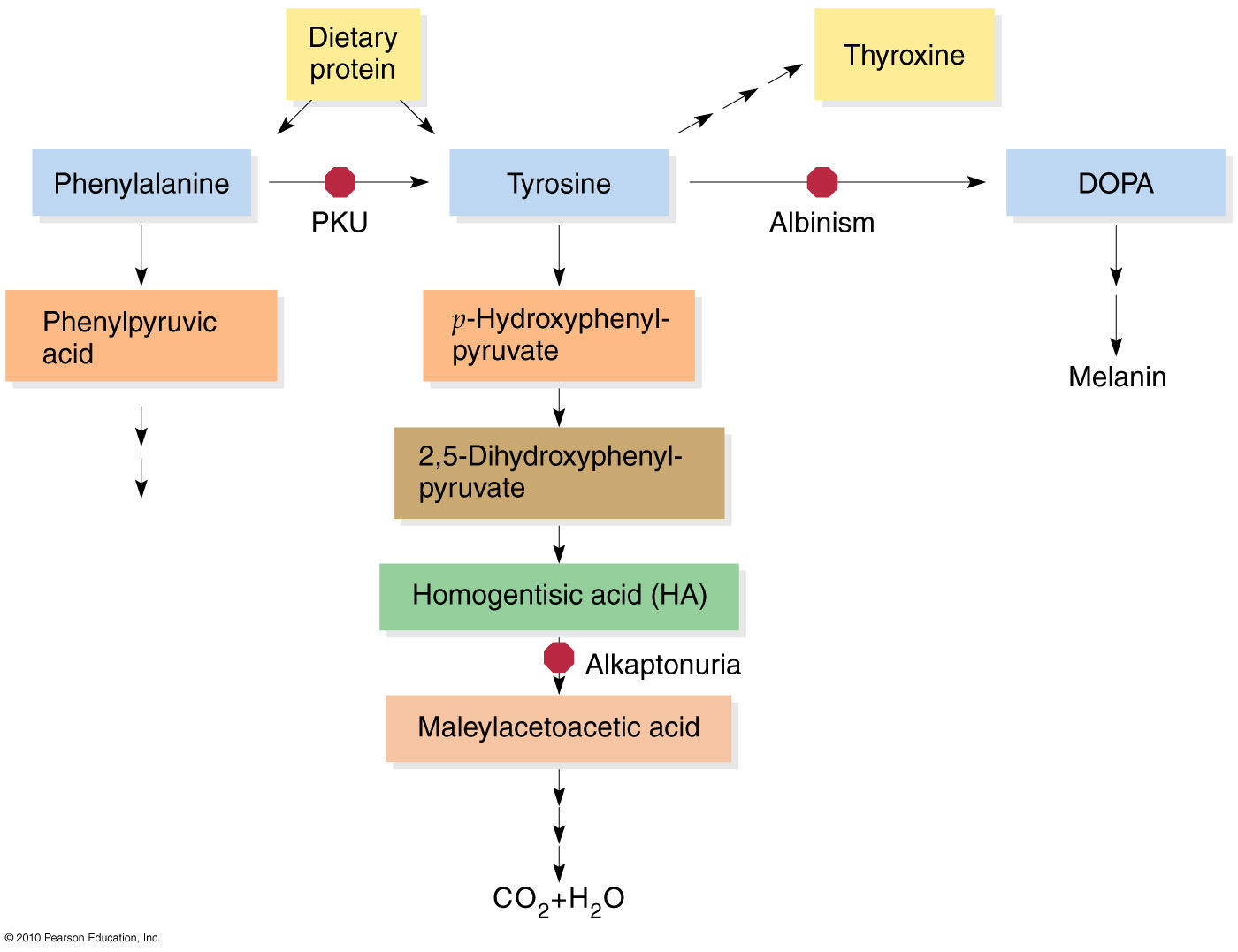
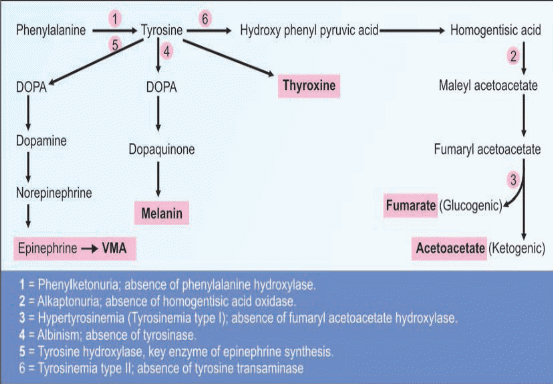
Abnormality in phenylalanine and Tyrosine metabolism
PHENYL KETONURIA (PKU)
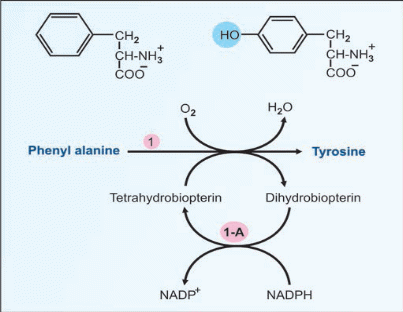
Deficiency of phenyl alanine hydroxylase (Fig.17.1) is the cause for this disease. The genetic mutation may be such that either the enzyme is not synthesized, or a non-functional enzyme is synthesized.
Phenyketonuria (PKU) is a genetic disorder that is characterized by an inability of the body to metabolize phenylalanine, caused by a deficiency in Phenylalanine Hydroxylase (PAH) enzyme or The defect is due to deficiency of dihydrobiopterin reductase.an enzyme that catalyzes the regeneration of tetrahydrobiopterin (cofactor of PAH)
4. Biochemical Abnormalities
A. Phenylalanine could not be converted to tyrosine. So phenylalanine accumulates. Phenylalanine level in blood is elevated.
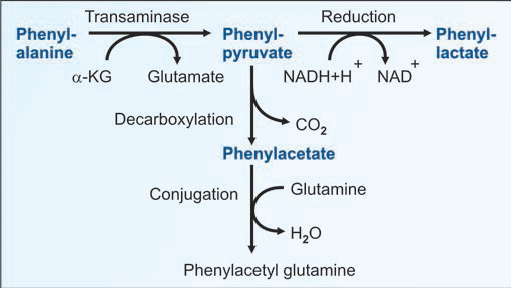
B. So alternate minor pathways are opened accumulation of too much phenylalanine and its toxic metabolities phenylpyruvic acid, phenyllactic acid and phenylacetic acid. which becomes a major donor of amino groups in aminotransferase activity and depletes neural tissue of α-ketoglutarate. Absence of α-ketoglutarate in the brain shuts down the TCA cycle and the associated production of aerobic energy, which is essential to normal brain development.
Phenyl ketone (phenyl pyruvate), phenyl lactate and phenyl acetate are excretedin urine.
Clinical Manifestations
A. The classical PKU child is mentally retarded
B. Agitation, hyperactivity, tremors and convulsions are often manifested. This may be because phenylalanineinterferes with neurotransmitter synthesis. Since tetrahydrobioptrerin is the co-enzyme
required for serotonin and dopamine, the decreased level of these neurotransmitters may also result in the neurological symptoms.
C. The child often has hypopigmentation, explained by the decreased level of tyrosine.
D. Phenyl lactic acid in sweat may lead to mousy body odor.
6. Laboratory Diagnosis
Blood phenylalanine: Normal level is 1 mg/dl. In PKU, the level is >20 mg/dl.
2-Tyrosinemia
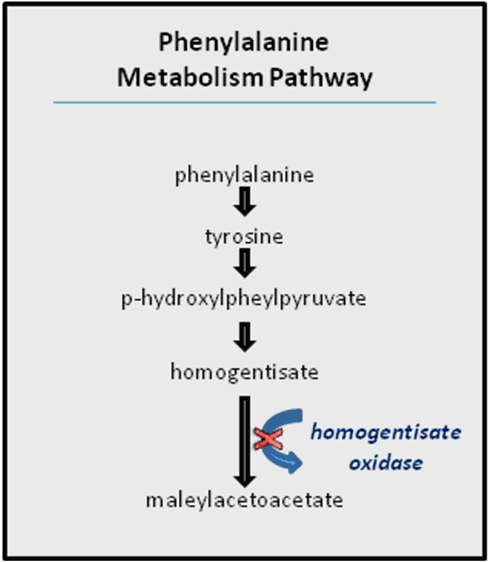
Tyrosine is an amino acid which is found in most animal and plant proteins. The metabolism of tyrosine in humans takes place in liver Tyrosinemia is caused by an absence of the enzyme fumarylacetoacetate hydrolase (FAH) which is essential in the metabolism of tyrosine. The absence of FAH leads to an accumulation of toxic metabolic products in various body tissues, which in turn results in progressive damage to the liver and kidneygiving a raised level of tyrosine in blood and urine clinical symptoms include moderate mental retardation, characteristic eye and skin lesions and disturbance in fine coordination.
3-Alkaptonuria (Black urine disease)
An inherited defect in the phenyl a lanine – tyrosine pathway involves a deficiency in the enzyme that catalyses the oxidation of homogentisic acid (an intermediate in the metabolic breakdown of tyrosine and phenyalanin).
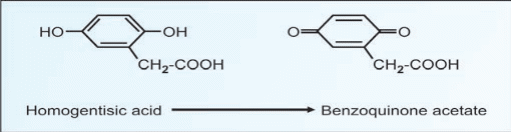
Alkaptonuria is caused by the lack of an enzyme called homogentisic dioxygenase (HGD). This condition occurs 1 in 1,000,000 live birth homogentisic acid accumulates and gets excreted in urine where the urine turns black on standing. There is a form of arthritis in late cases and generalized pigmentation of connective tissues; this is believed to be due to the oxidation of homogentisic acid by polyphenol oxidase
forming benzoquinone acetate that polymerises and binds to connects tissues molecules.High doses of ascorbic acid have been used in some patients, to help reduces the deposition of pigment on collagen, but progress of the disease has not been significantly affected by this strategy. Patients usually lead a normal life. It is compatible with fairly normal life. The only abnormality is the blackening of urine onstanding. The homogentisic acid is oxidized by polyphenol oxidase to benzoquinone acetate (Fig.17.6). It is then polymerized to black colored alkaptone bodies.
4-ALBINISM
1. The Greek word, albino means white. Albinismis an autosomal recessive disease with an incidence of 1 in 20,000 population (Fig.17.7).
Albinism – genetically determined lack or deficit of enzyme tyrosinase
Tyrosinase is completely absent, leading to defective synthesis of melanin.
3. The ocular fundus is hypopigmented and iris may be grey or red. There will be associated photophobia, nystagmus and decreased visualacuity.
4. The skin has low pigmentation, and so skin is sensitive to UV rays. The skin may show presence of naevi and melanomas. Hair is also white.
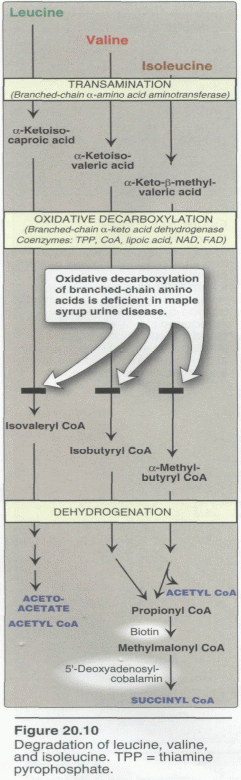
Branched chain amino acids Abnormality
Maple syrup urine disease (MSUD)
The normal metabolism of the branched chain amino acids Leucine, Isoleucine, and valine Valine. (Val) (V) is glucogenic; Leucine (Leu) (L)
is ketogenic while Isoleucine (Ile) (I) is both ketogenicand glucogenic. All the three are essential amino acids. Leucine is the major ketogenic amino acid.These amino acids serve as an alternate source of fuel for the brain especially under conditions of starvation.metabolism of these amino acid involves loss of the α-amino acid by transamination followed by oxidative decarboxylation of the respective keto acids. . The decarboxylation step is catalysed by branched chain α keto acid decarboxylase. In approximately 1 in 300,000 live birth in the general US population are affected by this enzyme defect leading to ketoaciduria. When untreated this condition may lead to both physical and mental retardation of the newborn and a distinct maple syrup odor of the urine.
This defect can be partially managed with a low protein or modified diet. In some instances,supplementation with high doses of thiamine pyrophosphate is recommended Thisis because the decarboxylation
requires thiamine..
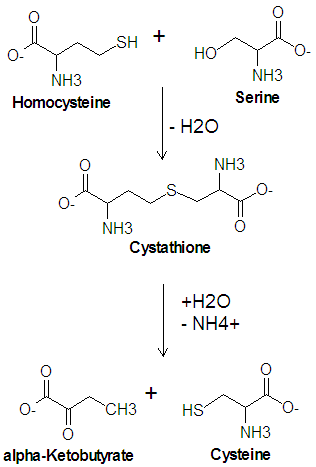
Homocystinuria
Norepinephrine
Cystinuria
Cystinuria is one of the inborn errors of metabolism. The disorder is attributed to
the deficiency in transport of amino acids is an inherited autosomal recessive disease that is characterized by the formation of cystine(cysteine-S-S cysteine) stones in the kidneys, ureter, and bladder. Cystinuria is a cause of persistent kidney stones. It is a disease involving the defective transepithelial transport of cystine and dibasic amino acids in the kidney and intestine, and is one of many causes of kidney stones.
1 NEONATAL RESPIRATORY DISTRESS INCLUDING CPAP CLINICAL LEARNING RESOURCE
1066 DEMENTIA RESEARCH GROUP PILOT STUDIES INDEPENDENT CLINICAL ASSESSMENT
10AP17] CLINICAL AND ENDOSCOPIC COMPARISON OF THE SINGLE USE
Tags: alsewedy inborn, biochemistry, alsewedy, clinical, stage, inborn, second, drthana, lecture
- CENTRAL BANK OF CYPRUS EUROSYSTEM ANNEX IV OF THE
- 15 ORDINANCE NO 9632 (NS) AN ORDINANCE ADDING CHAPTER
- HABÍA UNA VES DOS JÓVENES QUE VIVÍAN EN UN
- ZLINEU ZÁKLADNÍ INFORMACE KULTURA SPORT A
- EN ZIZUR MAYOR (NAVARRA) A OCHO DE JUNIO DE
- REAL DECRETO POR EL QUE SE CREAN LAS CATEGORÍAS
- DON XXXXXXXXX EMPLEOCUERPO DE LA ESCALA A EXTINGUIR DE
- PERIHAL BARU PERPANJANGAN ) SURAT PERMOHONAN PERPANJANGAN
- HOME SAFETY – GAS & CARBON MONOXIDE GAS CAN
- Declaracion Jurada Protocolo Aplicable al Covid 19 en Nuestro
- REPUBLIKA HRVATSKA KRAPINSKO – ZAGORSKA ŽUPANIJA Ž U P
- GUIA DE FORMULACIÓN DE PROYECTOS PROGRAMA + CONECTADOS ANILLOS
- 1) PASAPORTE ELECTRONICO ESTAMOS CONECTADOS CON EL SISTEMA RENAPER
- WITAJCIE! PONIŻEJ PODAJĘ WAM TEMATY LEKCJI I NOTATKI KTÓRE
- UNIDAD 1 CIENCIAS NATURALES LOS SERES VIVOS 13 ALUMNADO
- ROPA DNIA 27052014 ROKU (OZNACZENIE ZAMAWIAJĄCEGO) DO WSZYSTKICH
- AMBASSADE DE FRANCE EN ESPAGNE SERVICE DE PRESSE ET
- IASB EXPOSURE DRAFT – FINANCIAL INSTRUMENTS AMORTISED COST AND
- PSICOLOGÍA GENERAL PS VÍCTOR CABRERA VISTOSO ¿CUÁL ES EL
- AGREEMENT OF ADJECTIVES ANSWER SHEET 1 BONITA 26 VERDE
- LA MUESTRA ES UNA COPRODUCCIÓN ENTRE AMBAS INSTITUCIONES EL
- ANEXO 1 ENCUESTA SOBRE LA ENSEÑANZA ACTUAL DE LA
- 1 LA MEMBRANA PLASMÁTICA 11 CONCEPTO LA MEMBRANA PLASMÁTICA
- 62481 MATLAB USERS GUIDE MAY 1981 CLEVE MOLER DEPARTMENT
- THE EVERGREEN STATE COLLEGE SECTION 10100 LAB II CHEMISTRYGEOLOGY
- Advent Conspiracy can Christmas Still Change the World? Advent
- FACTSHEET LGBT+ MEMBERS IN UNISON – HOW TO GET
- „ROBINSON KRUZOE” – TEST ZE ZNAJOMOŚCI LEKTURY 1 Z
- ASESORAMIENTO PARA LA CREACIÓN Y PUESTA EN MARCHA DE
- CULTURA ORGANIZACIONAL SILVIA REGINA M DEL CIEL
CROSS COUNTRY SUMMER WORKOUT JUNE JULY AND AUGUST
ENTREVISTAS PLAZAS DE PROFESOR ASOCIADO RESOLUCIÓN 27062013 PÁGINA 1
DOCUMENT 1 GUIDE NOTES PUBLIC SECTOR TELEPHONE CONTACT CENTRE
SEDIMENTARY ROCK ACTIVITY PART I LOOKING AT SEDIMENTS SETTLING
POKLICNA MATURA V ŠOLSKEM LETU 20212022 POKLICNA MATURA JE
 WTW PARTICIPANT MONTHLY RECORD OF HOURS THIS RECORD IS
WTW PARTICIPANT MONTHLY RECORD OF HOURS THIS RECORD ISJUAN ANTONIO RODRÍGUEZ GARZA CORREO ELECTRÓNICO JUARODRIUQROOMX ESTUDIOS PROFESIONALES
NAPISATI SVAKI ZNAK 5 PUTA PORETKOM POTEZA U SKLADU
DIRECTORIO GENERAL DE MOVIMIENTOS ORGANISMOS Y ASOCIACIONES APOSTÒLICAS DE
 INFORMATIONEN ZUR ERHEBUNG PERSONENBEZOGENER DATEN 1 ZUR ERFÜLLUNG DER
INFORMATIONEN ZUR ERHEBUNG PERSONENBEZOGENER DATEN 1 ZUR ERFÜLLUNG DERWNIOSEK O AKREDYTACJĘ STAŻU KLINICZNEGO CZĘŚĆ I WNIOSEK MOGĄ
POLITICAL AND ECONOMIC POTENTIAL OF CHINA RESPECTED PROFESSOR DR
 SURGICAL BOOKING FORM ORPO100032G REV OCT 2013 PAGE 1
SURGICAL BOOKING FORM ORPO100032G REV OCT 2013 PAGE 1 ADVISORY COMMITTEES POLICYPROCEDURES OVERVIEW PURSUANT TO SECTION 451107 OF
ADVISORY COMMITTEES POLICYPROCEDURES OVERVIEW PURSUANT TO SECTION 451107 OFDEBER LENGUAJE TRIMESTRE I JIMMY MACIAS TEORÍA DEL
 POSITIVE FOR YOUTH VCS FUNDING – APPLICATION 201516 PLEASE
POSITIVE FOR YOUTH VCS FUNDING – APPLICATION 201516 PLEASE 7 AZ MSZ EN ACÉLJELÖLÉSI RENDSZER FELÉPÍTÉSE DR SZABADÍTS
7 AZ MSZ EN ACÉLJELÖLÉSI RENDSZER FELÉPÍTÉSE DR SZABADÍTS National Cyclone Risk Mitigation Project Kerala State Disaster Management
National Cyclone Risk Mitigation Project Kerala State Disaster Management SPECIFICATIONS CROWN SELF PRIMER PO8 PO10 WWWCRANEPUMPSCOM SCOPE FURNISH
SPECIFICATIONS CROWN SELF PRIMER PO8 PO10 WWWCRANEPUMPSCOM SCOPE FURNISHINTESTINAL DIGESTION KINETICS OF CASEIN OR MILK SOLUBLE PROTEINS
