TEHACHAPI VALLEY HEALTHCARE DISTRICT POLICY MEDSUN MEDICAL DEVICE
TEHACHAPI VALLEY HEALTHCARE DISTRICT POLICY MEDSUN MEDICAL DEVICE
|
POLICY: MedSun / Medical Device Reporting Policy |
POLICY NUMBER: 100.100 |
|
|
Original/Rewrite Approved: 12/28/2010 |
|
Originating Dept: Administration |
|
|
Applies to Depts: Clinical Departments |
|

POLICY
It shall be the policy of the Tehachapi Valley Healthcare District (TVHD) that all incidents involving patient care devices, related equipment hazards, and explant devices suspected of a possible failure be appropriately reported to the Safety Committee and MedSun (Medical product safety Network). This policy is required in order to conform to the Safe Medical Device Act of 1990 and FDA regulations.
PROCEDURE
This procedure applies to any personnel who discover, witness, or are notified of a suspected medical device malfunction, or problems such as close calls, or concerns regarding the safety of a device. Included within the scope of this policy are personnel who use or operate a medical device, including physicians, nurses, technicians, therapists, or other medical personnel.
A. Individual Reporting of Devices (Other than Impant/Explant Devices): Any individual who witnesses, discovers, or otherwise becomes aware of information that reasonably suggests that a medical device (equipment and/or product) has caused (or may cause) or contributed to the death, serious injury, or serious illness of a patient or employee of the facility, is responsible for immediately completing a Quality Review Report (see #)
In addition the following steps must be taken:
Take appropriate corrective action to address immediate safety/ operational/ treatment issues.
If the device is an electronic piece of equipment, record any measurement settings on the equipment controls. If possible, do not turn off equipment.
Collect all related disposables, packaging and accessories.
If the device is a disposable product, place product in a biohazard packaging.
Notify Maintenance Department. Tag defective equipment (See policy 110.39). .
The attending physician shall examine the patient's illness or injury related to the
incident, record the patient's physical findings, and document in the patient's progress notes the occurrence of the suspected adverse medical device incident and any actions taken based on the examination.
Departments Handling Implant/Explant Devices:
Implant/explant devices that are removed solely because of end of life and/or end of use are sent to Purchasing Department along with an explant form. The Purchasing Department will contact BioMedical and determine whether the device is trackable, send necessary notifications to the manufacturers, send the device to the manufacturer, return nontrackable devices to departments if requested, and/or dispose of the device.
Any individual who becomes aware of information that reasonably suggests that an implant/explant device has caused (or may cause) or contributed to the death, serious injury, or serious illness of a patient, is responsible for immediately completing a Quantros SRM Report.
The following information should be documented, if applicable:
What happened to the person affected
What were the problems with the device
What was the original medical procedure
What follow-up medical procedures were required because of the event
What type of device was involved, and what is the name of the manufacturer
What are the device identification numbers
How did the device cause or contribute to the event
The Risk Manager will log the event and notify the TVHD MedSun Representative. he TVHD MedSun Representative will, if appropriate, report the event via the MedSun system. It is the responsibility of the MedSun Representative to coordinate the educational process for all current and new employees and Medical Staff.
The Hospital Departmental Performance Improvement Committee, which reports to the Medical Staff PI Committee, and has the responsibility of implementing and managing the District’s medical device-reporting program. The Committee findings are confidential.
This responsibility will include the following:
Reviewing and analyzing all reportable incidents involving medical devices.
Reviewing and overseeing all reportable devices suspected of failure and/or
defect that might cause harm.
The Risk Management staff shall assist in coordinating reportable events and
oversee compliance for the Quantros Reporting and Adverse Event/
Sentinel Event policies. The Risk Management staff will be responsible for
the following activities:
Coordinate risk identification and investigation activities with appropriate
departments.
Ensure that all data collected from the medical device reporting program shall
be incorporated into the Quantros reporting system.
Review the recommendation of the Hospital Departmental Performance Improvement Committee for corrective actions
Involving any possible liability to the facility.
Purchasing Department is responsible for the procurement of medical devices and to coordinate with Biomedical Technology Services to investigate equipment product defects that are reported as incidents.
DEFINITIONS
MedSun: MedSun is an FDA internet-based system designed to provide a secure way to report adverse medical device events. MedSun also captures additional reporting on close calls or rejection of a device over safety concerns.
Safe Medical Device Act of 1990: The Safe Medical Device Act of 1990 was passed by the United States Congress to better protect the public health by increasing reports of device related adverse events by both manufacturers and user facilities.
Medical Device: In the Medical Device Act, the FDA defines a medical device as any
instrument, apparatus, or other article that is used to prevent, diagnose, mitigate, or treat a disease or to affect the structure or function of the body, with the exception of drugs. For example, a medical device includes but is not limited to ventilators, monitors, dialyzers, and any other electronic equipment, implants, thermometers, patient restraints, syringes, catheters, in vitro diagnostic test kits and reagents, disposables, components, parts, accessories, and related software. FDA Investigational Devices are not included in this policy. The principal investigator or their designee under stringent FDA guidelines handles these devices.
Implant/Explant Device: An implant/explant device is a device that is placed/removed
into/out of a surgically or naturally formed cavity in the human body. The purpose of this
device is to continually assist, restore, or replace the function of an organ system or
structure of the human body throughout the useful life of the device. These devices are
totally encased by the human body. Any external component must not form a physical
connection to the device.
Device Failure: A device failure is the failure of a device to perform or function as
intended, including any deviations from the device's performance specifications or
intended use.
FDA Reportable Event: A reportable event is an event where a device has the potential to cause or contributed to a serious injury, serious illness, or death of a patient or employee. This event may include equipment malfunctions and/or user errors. Reporting to the FDA is required within ten working days.
Reference:
Safe Medical Device Act of 1990 and 60 Fed Reg 63579 (1995)
Additional Policies:
Quantros, 100.28
Tagging Defective Equipment, 110.39
Tags: device reporting, medical device, district, healthcare, policy, valley, tehachapi, device, medical, medsun
- ORACIÓN ANTE UN NUEVO AÑO POR JAVIER LEOZ SEÑOR
- FSCTPL01002 APPLICATION FOR A DEROGATION TO USE A HIGHLY
- INFORMACIJSKO SREDIŠČE PUBLIKACIJE STATISTIČNEGA URADA IZDANE OD 23 DECEMBRA
- THIRD PRESBYTERIAN CHURCH DEACONS’ MEETING FEBRUARY 23 2021 LYNDA
- VIENTO SUR NÚMERO 88SEPTIEMBRE 2006 UNA CRÍTICA FEMINISTA A
- EJERCICIOS SOBRE PROBABILIDADES UCA – DPT MATEMÁTICA 1 SE
- P AFZAL ET AL JMM 48 A (1)
- STI CONFERENCE 2018 · LEIDEN TEMPLATE FOR SUBMISSION OF
- 20172018AS TANÉV 1 A BETÖLTÖTT MUNKAKÖRÖK ALAPJÁN A PEDAGÓGUSOK
- CON LAW OUTLINE RULES AND ANALYSIS I READ THIS
- MARIA VIRGEN Y MADRE OFRECEMOS ESTA CELEBRACIÓN PARA LA
- TUSCARORA BLENDED LEARNING CHARTER SCHOOL THE SUCCESSFUL ONLINE STUDENT
- FOOD PROCESSING IN WEST BENGAL IN FOOD AND AGRO
- DOCKET NO 361 FINDINGS OF FACT PAGE 15 DOCKET
- AUTOMATA MIATT TUDOK PAR TANACSOT ADNI DONGESD MEG JOL
- PODACI O RAJONIMA ZA OBAVLJANJE DIMNJAČARSKIH POSLOVA NA PODRUČJU
- LEY 819 09072003 DIARIO OFICIAL 45243 POR LA CUAL
- 5) LA LISTE DES COURS (SIGLE ET TITRE) QUE
- MATHS TEST PAPER MEASUREMENT OF TIME AND LENGTH TT
- BOOKING FORM TOOLBOX TALK NO 4 – REVIEWING A
- AYUNTAMIENTO DE COLMENAR (MÁLAGA) ORGANIGRAMA DE LA
- POUR LA VERSION FRANÇAISE CLIQUEZ ICI THE AFRICAN UNION
- SOLICITUD DE RECTIFICACIÓN DE LA AUTOLIQUIDACIÓN Y DE DEVOLUCIÓN
- NEW FEATURES IN OC RDC 51 RDC CLASSIC
- UNIVERSIDAD CENTROCCIDENTAL “LISANDRO ALVARADO” COORDINACIÒN DE PROTOCOLO
- ANÁLISIS DE SEGREGACIÓN DE LOS XSTRS DEL DECAPLEX NOMBRE
- MÅNEDSBREV FRA VEPSEBOLET OG MAURTUEN JUNI 2017 TILBAKEBLIKK FRA
- EU VET PROGRAMME II BOSNIA AND HERZEGOVINA FAMILIJA SVE
- BILAGA 3 CHECKLISTA FÖR TILLSYN HOS ÅTERFÖRSÄLJARE AV
- CEDAR HILL GIRLS SOFTBALL ASSOCIATION PLAYER COMPETENCY EXPECTATIONS BY
2 PHYSICAL SCIENCE WORKSHOP ASTRONOMY APPLICATIONS OF LIGHT &
OPIS EGZAMINU MATURALNEGO Z JĘZYKA POLSKIEGO 2014 EGZAMIN MATURALNY
{0CASH ASSISTANCE SEVERANCE BENEFIT AGREEMENT}0{ACUERDO DE INDEMNIZACIÓN POR FIN
 CHEROKEE ADVOCATE JUNE 18 1870 GROUND COUNCIL OF INDIANS
CHEROKEE ADVOCATE JUNE 18 1870 GROUND COUNCIL OF INDIANSTEMİZLİK ARAÇLARI VE GEREÇLERİ TEMİZLİK KİMYASALLARI KIRTASİYE SARF MALZEMELERİ
APOYO A PROYECTOS DE DESARROLLO Y ACELERACIÓN DE TECNOLOGÍAS
 DEPARTAMENTO DE CIENCIAS PROFESORA PATRICIA ORIA M MAIL PATYYOMHOTMAILCOM
DEPARTAMENTO DE CIENCIAS PROFESORA PATRICIA ORIA M MAIL PATYYOMHOTMAILCOMSEÑORES DIRECCIÓN SECCIONAL DE ADUANAS DE DIRECCIONES DE
 REGISTER OF INTEREST INFORMATION SHEETBOARDSCOMMITTEESCOUNCILS I THE UNDERSIGNED (SURNAME
REGISTER OF INTEREST INFORMATION SHEETBOARDSCOMMITTEESCOUNCILS I THE UNDERSIGNED (SURNAMEÐïࡱáþÿ Þÿÿÿ !&()+0123456789?[]^`
 ПРИЛОГ БР ПРОЦЕДУРА ХИТНИХ СТАЊА КАБИНЕТА ЗА НЕУРОПСИХИЈАТРИЈУ БРОЈ
ПРИЛОГ БР ПРОЦЕДУРА ХИТНИХ СТАЊА КАБИНЕТА ЗА НЕУРОПСИХИЈАТРИЈУ БРОЈLUIS DE GÓNGORA – SONETO CLXVI MIENTRAS POR COMPETIR
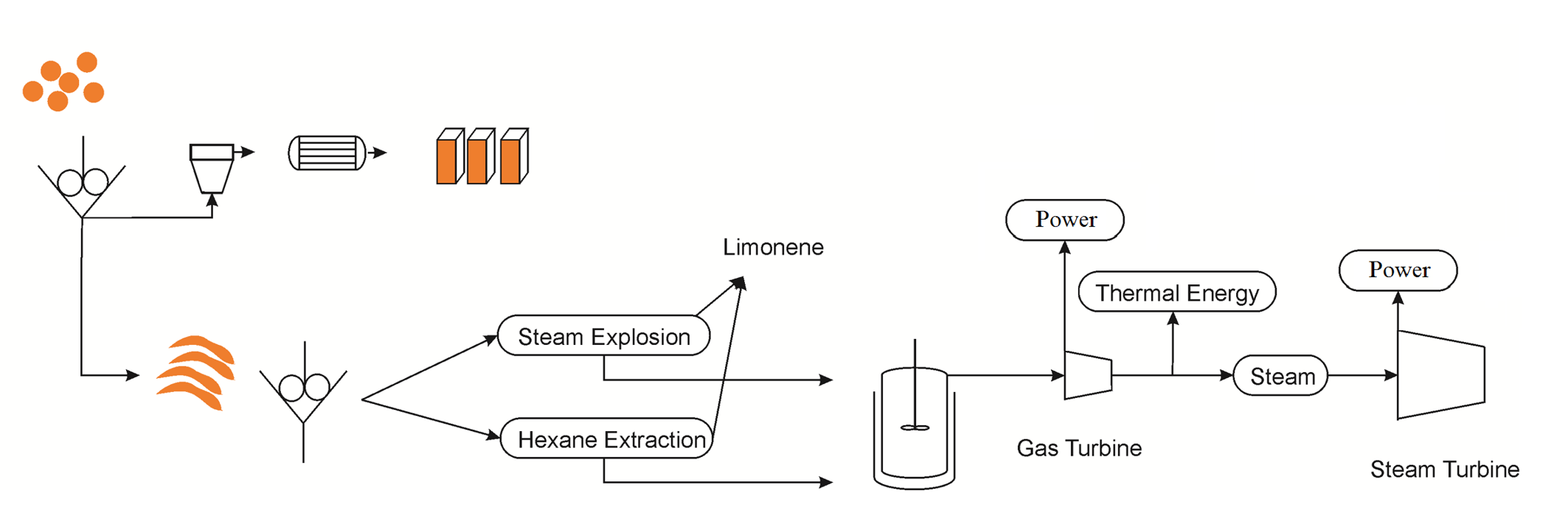 INTEGRATED MULTIPRODUCT FACILITY FOR THE PRODUCTION OF CHEMICALS FOOD
INTEGRATED MULTIPRODUCT FACILITY FOR THE PRODUCTION OF CHEMICALS FOOD2547 SAYILI YÜKSEK ÖĞRETİM KANUNU’NA GÖRE MESLEK YÜKSEKOKULLARINDA BİLİNMESİ
 TŘÍDNÍ VZDĚLÁVACÍ PROGRAM MATEŘSKÉ ŠKOLY SVĚT PATŘÍ VŠEM“ MATEŘSKÁ
TŘÍDNÍ VZDĚLÁVACÍ PROGRAM MATEŘSKÉ ŠKOLY SVĚT PATŘÍ VŠEM“ MATEŘSKÁ MINISTERO DELLA PUBBLICA ISTRUZIONE UFFICIO SCOLASTICO REGIONALE PER LA
MINISTERO DELLA PUBBLICA ISTRUZIONE UFFICIO SCOLASTICO REGIONALE PER LA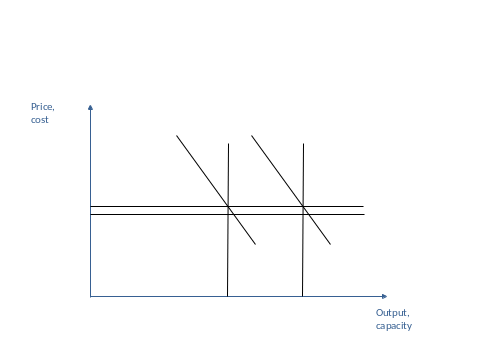 17 INFRASTRUCTURE REGULATION AND INVESTMENTS PETER FORSYTH DEPARTMENT OF
17 INFRASTRUCTURE REGULATION AND INVESTMENTS PETER FORSYTH DEPARTMENT OF(SPONSORING CO T2)COPY ONTO THE PASS SPONSORING COMPANY HEADED
 ASSIGNMENT 2 OF SUPERVISED LEARNING NAME NOTE THIS QUESTION
ASSIGNMENT 2 OF SUPERVISED LEARNING NAME NOTE THIS QUESTION DIRECTRIZ ADMINISTRATIVA DRPI062012 DE LIC LUIS GUSTAVO ÁLVAREZ RAMÍREZ
DIRECTRIZ ADMINISTRATIVA DRPI062012 DE LIC LUIS GUSTAVO ÁLVAREZ RAMÍREZ
