PATOLOGÍA MITOCONDRIAL BQ21 DR GARESSE LA PATOLOGÍA MITOCONDRIAL TIENE
PATOLOGÍA MITOCONDRIAL BQ21 Dr
PATOLOGÍA MITOCONDRIAL BQ21 Dr. Garesse
La patología mitocondrial tiene una variabilidad fenotípica enorme. Dependiendo de la vía metabólica que se altere dará unos u otros síntomas: diabetes, encefalomiopatía devastadora... (antes eran conocidas como enfermedades raras”). Realmente no son tan extrañas debido al aumento de su conocimiento: 3-5 / 10.000hab. Se diagnostica mal, porque los síntomas son difusos y la evolución es impredecible. Puede aparecer a cualquier edad, con cualquier tipo de herencia, y pueden existir varios individuos de una familia afectos.
No todas las mc son iguales; existen muchos tipos en función del tipo celular en el que se encuentren: astrositos, músculo cardiaco, glándula SR, gl pineal,…
La mitocondira (mc) está presente en todas las células del organismo (enfermedad metabólica más frecuente) y tiene una gran variabilidad funcional:
oxidación ác grasos
ciclo urea
ciclo ác tricarboxílicos
metab mtDNA
elongación ác grasos
desaturación ác grasos
síntesis fosfolípidos
MAO
Pero la función más importante es la síntesis de ATP por medio de la cadena respiratoria.
La patología mitocondrial en teoría abarcaría muchos más procesos que las alteraciones específicas de la fosforilación oxidativa, pero en la práctica, éstas constituyen el 99% de la patología mc. Así, la patología mc es sinónimo de enfermedad de la cadena respiratoria e implica una disminución de la síntesis de ATP. Como todo el organismo funciona gracias al ATP, sólo pueden concebirse déficits parciales de la síntesis de ATP, porque un déficit total sería incompatible con la vida.
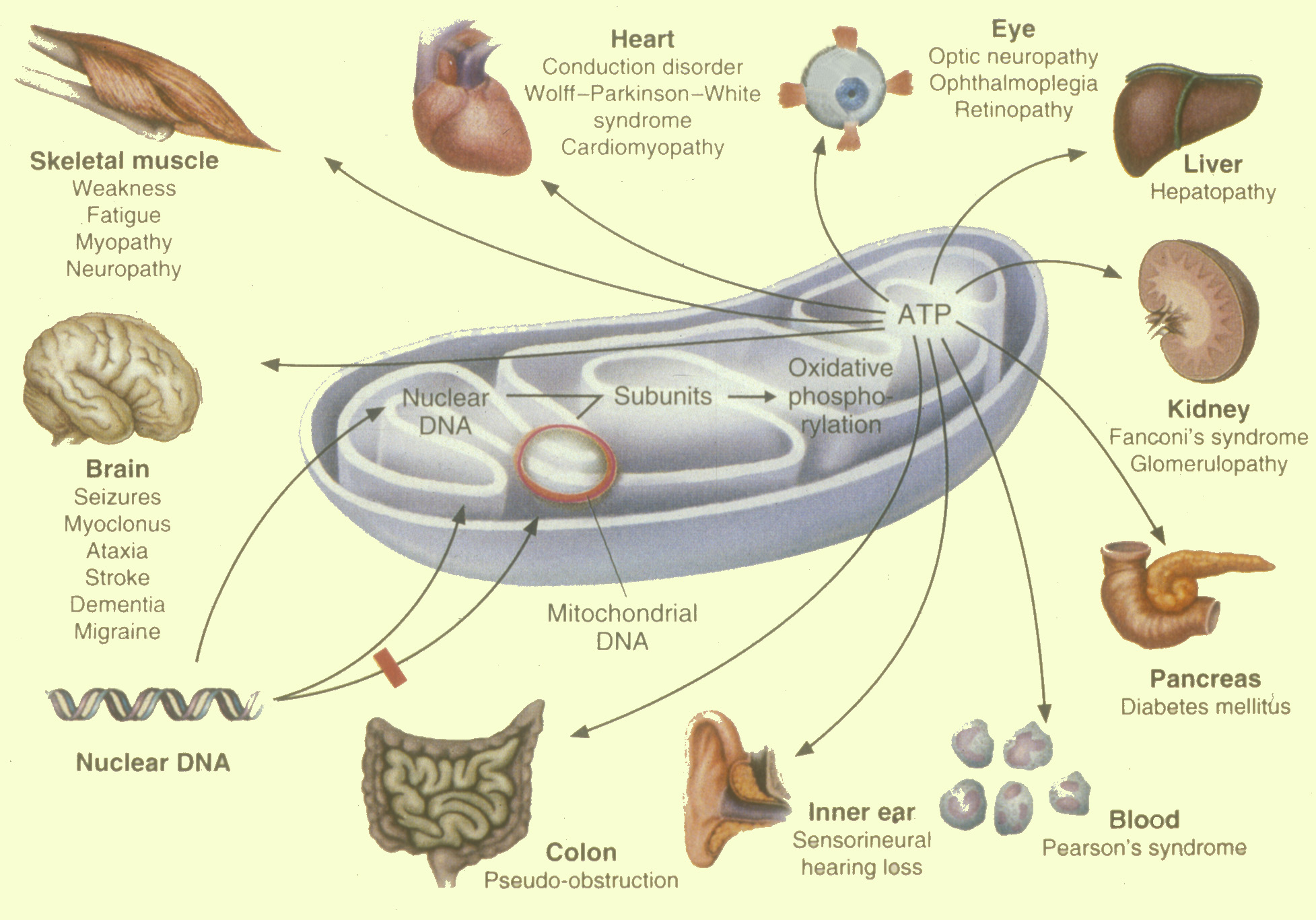
Estas patologías son de afectación multisistémica. La manifestación clínica puede ser la afectación de un solo órgano (sordera, cardiomiopatía...) o una combinación de síntomas (generalmente encefalomiopatías con otros). Afectan más a músculo y SNC porque estos órganos tienen un metabolismo oxidativo elevado (cuanto mayor consumo de ATP mayor afectación).
CADENA RESPIRATORIA
Hay 5 complejos que recogen la energía en forma de NADH (es el poder reductor de esta molécula lo que se acumula) por medio de un gradiente de protones lleva a la síntesis de ATP. Cualquier mutación o defecto implica un déficit de la síntesis de ATP. Hay 70 proteínas distintas que se ocupan de la síntesis de ATP, es decir, 70 posibilidades de fallo.
GENÉTICA MITOCONDRIAL
Papel central de la mc en la fisiología celular:
-Apoptosis
-Envejecimiento
-Desarrollo
-Diferenciación celular
-Regulación nivel calcio intracelular
-Proliferación celular
Biogénesis mitocondrial:
-Proliferación celular
-Diferenciación celular (ej. Músculo cardíaco posee mc completamente desarrolladas, llenas de crestas).
DNA mitocondrial
La mc se diferencia del resto de las organelas celulares en que contiene DNA propio: es pequeño, de unos 16.500 bp (DNA nuclear tiene 9.3 109-10). Cada mc posee de 2 a 10 moléculas de DNA. Teniendo en cuenta que cada célula tiene miles de mc, la cantidad de mtDNA es relativamente importante. Es un DNA pequeño, pero muy repetido.
En el patrón de herencia de la cadena respiratoria intervienen 2 sistemas genéticos: el DNA nuclear y el mitocondrial.
DNAnuclear: “mar” de nucleótidos con pocas secuencias codificantes.
DNAmt, prácticamente igual para todos los animales:
Pequeña región no codificante (1Kb): elementos reguladores de la replicación y la expresión. (El resto del genoma es codificante.)
Genes de 3 tipos:
- 2 rRNA de 12S y 16S que codifican para ribosomas de la mc. Es una estructura muy compacta (hay genes solapados).
- 22 tRNA de la síntesis de proteínas mitocondriales.
- 13 proteínas, subunidades de la cadena respiratoria: 7 subunidades del complejo I (ND1-ND7), 3 subunidades del complejo IV: la citocromo oxidasa (CO I, II, III), 1 subunidad del complejo III (citocromo b) y 2 subunidades de la ATP sintetasa (ATPasa 6 y 8).
El material genético restante para sintetizar ATP está en el núcleo.
El código genético del mtDNA es distinto del código del DNA nuclear.
La herencia del mtDNA es materna y el único mecanismo de evolución y variación de la secuencia del mtDNA es mediante la acumulación secuencial de mutaciones y su dispersión en la línea materna. La capacidad codificante del mtDNA es pequeña pero fundamental para la fisiología celular.
El mtDNA tiene una elevada velocidad de mutación debido a la falta de histonas (estando el DNA sin protección), a la continua generación de radicales libres en la mc como subproducto de la respiración, y a la falta de mecanismos de reparación.
Resumen mtDNA:
molécula circular covalentemente cerrada
nº copias elevado (poliploide)
herencia materna
organización muy compacta
código genético propio
semiautónomo
codifica únicamente subunidades OXPHOS (de la cadena respiratoria)
Origen del mtDNA
Las mc son resultado de una endosimbiosis: una bacteria aerobia colonizó una bacteria anaerobia y convivieron las dos. Cada una tenía su DNA, un DNA quedó como nuclear y el otro como mitocondrial. Con el tiempo mtDNA fue transfiriendo su DNA al núcleo, pero quedaron 13 genes (los correspondientes a las proteínas de la cadena respiratoria) sin transferir. Se mantienen estas 13 proteínas mitocondriales porque le son imprescindibles a la célula: la síntesis de ATP y, por tanto, la supervivencia celular, dependen de la cadena de transferencia de electrones, que requieren para su funcionamiento estas 13 proteínas.
A la célula le cuesta mucho gasto de síntesis de proteínas y energía la síntesis de estas 13 proteínas: tiene que replicar, transcribir y regular este mtDNA por medio de proteínas que se sintetizan en el núcleo y penetran en la mc (la replicación, transcripción,... de las proteínas mitocondriales se produce en la mc). MtDNA es, por tanto, semiautónomo.
MtDNA interviene en la síntesis de todos los complejos de la cadena respiratoria, excepto en la del complejo II. Hay muchas más subunidades codificadas por el DNA nuclear. Hay unas 60 mutaciones responsables del fallo de la cadena respiratoria: más de 50 son por mutaciones en mtDNA, menos de 10 por mutaciones en el DNA nuclear.
Al ser el código genético del mtDNA diferente al del núcleo, no le sirven las mismas proteínas de transcripción,... que se usan para las funciones del núcleo.
Cada célula tiene un número y morfología variable de mc, de acuerdo a sus necesidades de ATP.
Herencia del mtDNA
Todos tenemos las mismas moléculas de mtDNA pero entre nosotros hay diferencias: polimorfismos. La herencia es materna pura (ver anexo al final), el padre no transmite su mtDNA a sus hijos:
el espermatozoide contiene bastantes mc (las requiere para moverse), sin embargo en el momento de la fecundación, al producirse la transferencia de pronúcleos, pasan muy pocas,y las que se transfieren son marcadas con ubiquitina para ser destruídas. De esta manera las mc paternas se diluyen.
El ovocito tiene gran cantidad de mc (si normalmente una célula tiene del orden de 1000 a 2000 mc, el ovocito tiene de 100.000 a 200.000). El mtDNA es el prioritario del ovocito.
Sin embargo, tanto hombres como mujeres pueden padecer estas enfermedades.
Hipótesis de la Madre Eva
La secuencia del mtDNA es polimórfica: se puede estudiar cómo se propagan las razas analizando las variaciones del mtDNA. Así se reconstruye el árbol genealógico.
Cuando aparece una mutación en un gen nuclear en la célula germinal, pasa al resto de las células, pero cuando muta una molécula de mtDNA, al haber 100.000 copias, se diluye porque, ¿qué es una mutación en 100.000 copias?
Durante el proceso de oogénesis hay sólo unas pocas mc que se multiplican. Es muy difícil que cuando se muta una mc todos los mtDNA estén igualmente mutados. Posteriormente, al dividirse y multiplicarse habrá:
células con mtDNA mutado: homoplasmia con mutación
células con mtDNA sano: homoplasmia salvaje
células con mtDNA con y sin mutación: heteroplasmia
División celular → líneas diferentes → mutadas
↘ no mutadas
Célula con 1 mutación en mtDNA
↓
división, reparto de mc y nueva expansión
↙ ↓ ↘
cél mtDNA sano cél mc mutadas y sanas cél mc mutadas
(homoplasmia (heteroplasmia) (homoplasmia con
salvaje) mutación)
El mtDNA normalmenta se transmite en heteroplasmia y da lugar a un sujeto mosaico (unas células con homoplasmia y otras con heteroplasmia). Además, existe variabilidad en la trasmisión: dos hijos afectos de la misma madre pueden presentar distinta gravedad al tener también distinto grado de heteroplasmia.
Umbral de expresión fenotípica: La herencia de mc mutadas es al azar, no se sabe qué células reciben las mc mutadas: según a qué tejidos y el requerimiento de ATP de dicho tejido, expresarán más o menos clínica. Cerebro y músculo son los más afectados porque las necesidades de ATP son elevadas, con que fallen un poco sus mc y se sintetice menos ATP de lo normal, ya aparece patología: bajo umbral de patología (cada tejido tiene un umbral de patología diferente).
Así, dentro de una misma familia un individuo puede estar más afectado que otro porque:
cada uno tiene un porcentaje de mc mutadas, según cuántas mutaciones le haya transmitido la madre.
- Según la distribución de las mc mutadas en los tejidos.
Hay que recordar que la enfermedad es multisistémica, aunque la clínica esté dominada por la encefalomiopatía. Puede presentarse aisladamente una cardiomiopatía e ir avanzando a patología renal, ocular...
Las manifestaciones pediátricas son más graves. En el adulto en cambio puede debutar más tardíamente a partir de una intolerancia al ejercicio con deterioro progresivo conforme envejece -con la edad las mc van perdiendo eficacia, porque con la respiración se acumulan radicales libres que dañan las mc- dando epilepsia, debilidad muscular, ataxia, etc. Hasta que no se llega al umbral de patología no suelen aparecer los síntomas.
Puede ser que se transmitan únicamente en homoplasmia de generación en generación, porque al ser enfermedades poco agresivas sobreviven los enfermos como para tener descendencia.
El fenotipo que origina una mutación en el genoma mitocondrial depende finalmente de tres factores fundamentales:
El tipo de mutación
La cantidad de moléculas que llevan la mutación
La diferente demanda respiratoria de los tejidos, siendo mayor en músculo, sistema nervioso, riñón, páncreas, hígado.
Un cuarto factor lo constituye la edad del individuo afectado
C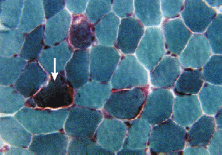 aracterísticas
de la patología mitocondrial:
aracterísticas
de la patología mitocondrial:
Esporádica, de herencia materna o mendeliana
Afectación específica o multisistémica
Variabilidad fenotípica en miembros de una misma familia
Aparición de los síntomas a cualquier edad
E volución
progresiva de la sintomatología
volución
progresiva de la sintomatología
Relación desconocida entre genotipo y fenotipo
DIAGNÓSTICO
B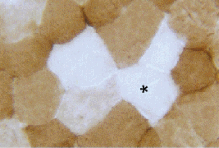 iopsia
muscular, porque suele acabar afectando al músculo.
Aparecen fibras rojo rasgadas. Con
tinción tricrómico
de Gomoy se ven
las membranas de las mc que han intentado proliferar para suplir el
déficit de ATP de color rojo fuerte alrededor de la célula
(en la periferia).
iopsia
muscular, porque suele acabar afectando al músculo.
Aparecen fibras rojo rasgadas. Con
tinción tricrómico
de Gomoy se ven
las membranas de las mc que han intentado proliferar para suplir el
déficit de ATP de color rojo fuerte alrededor de la célula
(en la periferia).
T inción
de citocromo oxidasa (que es el complejo IV de la
cadena respiratoria). Normalmente
las mitocondrias son de color marrón, pero al fallar la
citocromo oxidasa (COX) en las mc mutadas habrá una gama de
colores, según la intensidad de la afectación.
inción
de citocromo oxidasa (que es el complejo IV de la
cadena respiratoria). Normalmente
las mitocondrias son de color marrón, pero al fallar la
citocromo oxidasa (COX) en las mc mutadas habrá una gama de
colores, según la intensidad de la afectación.
M
 edida
de la actividad enzimática de la cadena respiratoria.
edida
de la actividad enzimática de la cadena respiratoria.
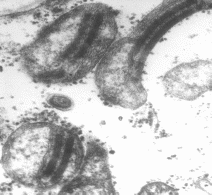
D esestructuración
de la mc al microscopio electrónico.
esestructuración
de la mc al microscopio electrónico.
Síntomas difusos. Hay un deterioro progresivo rápido. Se afecta un tejido (apareciendo DM, fallo hepático, tubulopatía renal) o afecta progresivamente al cerebro o al músculo. Los síntomas más frecuentes son: convulsiones, ataxia, debilidad muscular, distonía, demencia, miopatía.
En realidad, el ojo clínico, la afectación de distintos órganos y las pruebas funcionales del SNC y músculo dan el diagnóstico de sospecha. Puede hacerse un diagnóstico molecular si se ve la mutación en el mtDNA (mediante extracción del DNA de músculo o de células sanguíneas) o si existe una alteración en la cadena respiratoria.
Realizar estudio familiar.
La biopsia muscular y la tinción de COX permiten el diagnóstico histopatológico.
PATOLOGÍA MITOCONDRIAL
Puede ser por alteración del genoma nuclear (patrón de herencia mendeliano) o mitocondrial.
La patología molecular más frecuente es la mitocondrial (aunque individualmente cada patología no es muy frecuente, en conjunto, al ser muchas, sí resulta un número alto).
 Diferentes
alteraciones en tRNA pueden dar la misma patología (al fin y
al cabo falla la cadena respiratoria) y mutaciones parecidas en tRNA
pueden dar clínica diferente. Pero existen mutaciones que dan
preferentemente la misma patología. Ej. MELAS (Encefalopatía
Mitocondrial con Acidosis Láctica y Strokes) en 80%
tiene una mutación en 3243 y MERFF (Epilepsia Mioclónica
con Fibras Rojo Rasgadas) en la mayoría tiene la mutación
8344. Lo que no se conoce es por qué un genotipo produce un
fenotipo característico.
Diferentes
alteraciones en tRNA pueden dar la misma patología (al fin y
al cabo falla la cadena respiratoria) y mutaciones parecidas en tRNA
pueden dar clínica diferente. Pero existen mutaciones que dan
preferentemente la misma patología. Ej. MELAS (Encefalopatía
Mitocondrial con Acidosis Láctica y Strokes) en 80%
tiene una mutación en 3243 y MERFF (Epilepsia Mioclónica
con Fibras Rojo Rasgadas) en la mayoría tiene la mutación
8344. Lo que no se conoce es por qué un genotipo produce un
fenotipo característico.
 Alteración
del sistema OXPHOS da patología multisitémica:
Alteración
del sistema OXPHOS da patología multisitémica:
Mutaciones en el DNA nuclear
Aunque de las más de 100 subunidades diferentes que forman parte de los complejos respiratorios sólo 13 están codificadas en el genoma mitocondrial, la mayoría de los síndromes provocados por una disminución en la síntesis de ATP que se han caracterizado molecularmente están causados por alteraciones del genoma mitocondrial, aunque en el último año se han identificado varias mutaciones en genes nucleares que provocan una disfunción de la cadena respiratoria mitocondrial.
Fallos en el mtDNA con herencia nuclear: existen delecciones múltiples o depleciones. Puede deberse a que fallen proteínas que se codifican en el núcleo, pero como influyen en el control del mtDNA da la misma clínica que si fallase este mtDNA.
Puede haber fallos en la cantidad o en la calidad del mtDNA.
ANEXO
La intolerancia al ejercicio es una de las patologías mitocondriales, por mutaciones en el complejo III (citocromo b). Ya hemos dicho que todas estas patologías son de muy reciente descubrimiento, tanto es así que hace un mes, estudiando la intolerancia al ejercicio, se ha visto una mutación nueva esporádica. Se haplotipó el DNA del sujeto afecto y el mtDNA de la madre (puesto que la herencia es materna) y se vio que el haplotipo era el del padre (el mtDNA del padre segregó al hijo). Es más, el 100% de las moléculas del padre estaban en el músculo (por eso intolerancia al ejercicio). ¿Qué es lo que ocurrió? ¿Fallo en el mecanismo de eliminación de mc paternas? ¿Quedan susceptibles a mutaciones? Muchas preguntas de momento sin respuesta.
Polimorfismo del mtDNA
La variación genética del mtDNA es el producto de mutaciones que se han ido acumulando a lo largo de la evolución y de las que han surgido en tiempos más recientes. La mayoría de las variaciones son sustituciones neutras (en la tercera posición de los codones y en la región no codificante). Sin embargo, algunos afectan a aminoácidos medianamente conservados, que pueden provocar una pequeña disminución en la capacidad oxidativa celular. Si bien estas mutaciones no originan una patología por sí misma, pueden potenciar el efecto de otras mutaciones.
Los casos más claros de este efecto se han descrito en pacientes con LHON. También entrarían dentro de este grupo mutaciones que podrían ser importantes en la aparición de ciertas enfermedades degenerativas durante la vejez.
A medida que la mutación afecta a posiciones funcionalmente más importantes, la gravedad de los síntomas aumenta. Las más graves, que conllevan una importante disfunción, se eliminan rápidamente mediante selección natural. Por ello las mutaciones graves son recientes y a menudo son heteroplásmicas.
Mutaciones moderadamente deletéreas son, por ej., MTND1 LHON 3460A; MTND4 LHON 11778A; MTTK MERRF 8344A; MTTL1 MELAS 3243C. Los individuos que llevan estas mutaciones, incluso en elevadas proporciones, en general presentan pocos síntomas durante la juventud, y la patología se manifiesta durante la etapa adulta.
Las mutaciones gravemente deletéreas por el contrario, provocan síntomas rápidamente, afectando, por tanto, a la capacidad reproductiva del individuo, y las líneas maternas por tanto desaparecen rápidamente. Por tanto los casos de familias independientes descritos son nuevos, manifestando una variabilidad clínica muy grande dentro de la misma debido a la segregación replicativa. Entre ellas se encuentran: MTND6 LDYT14459A (LHON y distonía); MTATP6 NARP 8993G/C; MTTL1 MMC 3303T; MTTP MM 15990A.
PATOLOGÍA MITOCONDRIAL
(Cadena respiratoria)
MUTACIONES EN EL mtDNA
Reorganizaciones. Generalmente esporádicas.
Oftalmoplejía Externa Progresiva Crónica (CPEO)
Síndrome de Kearns-Sayre
Diabetes y sordera de herencia materna
Mutaciones puntuales (>50). Herencia materna.
En genes de proteínas
Neuropatía Óptica Hereditarie de Leber (LHON)
Neuropatía, Ataxia y Retinitis Pigmentosa (NARP)
Síndrome de Leigh de herencia materna (MILS)
En genes de tRNAs
Encefalomiopatía Mitocondrial con Acidosis Láctica y Strokes (MELAS)
Epilepsia Mioclónica con Fibras Rojo- Rasgadas (MERRF)
Sordera neurosensitiva
En genes de rRNAs
Sordera inducida por aminoglucósidos
Delecciones / Duplicaciones: son multisistémicos: retinopatía siempre, y además cardiopatía, sistema hematopoyético, páncreas.
Mutaciones puntuales: sintomatología muy variada: cada tipo de mutación enf. con cuadro clínico diferente. Delecciones similares cuadros diferentes (no existe relación clara entre genotipo y fenotipo). Distintas mutaciones mismo cuadro.
LHON: nervio óptico afectado pérdida de visión periférica (ceguera). Muchos son homoplásmicos.
MELAS: episodios de infarto cerebral, parálisis.
Si afectación rRNA sordera.
MUTACIONES EN GENES NUCLEARES
Proteínas estructurales de la cadena respiratoria (= sistema OXPHOS)
Complejo I: NDUFS 4, 7, 8:Síndrome de Leigh
NDUFS 2: Encefalomiopatía y cardiomiopatía hipertrófica.
Complejo II: Flavoproteína Síndrome de Leigh
Genes no estructurales
Ensamblaje complejo III:
BCS1L Tubulopatía y encefalopatía
Complejo IV:
Surf-1 Síndrome de Leigh
SCO-2 Cardioencefalomiopatía
SCO-1 Encefalopatía / Fallo hapático
COX-10 Síndrome de Leigh and De Toni-Fanconi-Debre
Homeostasis e Importe
Frataxina Ataxia de Friedreich
Paraplegina Paraplejía Espástica Hereditaria
DDP Síndrome de sordera-distonía
OPA-1 Atrofia Óptica Dominante
Comunicación Intergenómica
Delecciones múltiples: ANT1, PolG, helicasa
Depleción: dGK, TK
MNGIE: TP
Complejo IV: genes de regulación de transporte de proteínas al interior de la mc.
Comunicación intergenómica:
ANT-1 saca ATP de la mc.
Timidina fosforilasa interviene en el metabolismo de los nucleótidos.
Encefalomiopatías u oftalmoplejías externas con transmisión mendeliana. Delecciones múltiples en el núcleo en genes que regulan la homeostasis del mtDNA.
Ideas importantes:
Patología difícil de dgcar por su variablidad fenotípica y por su peculiar herencia propia.
Es la metabolopatía más frecuente.
A continuación algunas cosillas que venían en la presentación pero que no dio tiempo en clase y a las que
SD de Leigh: NO CLASE
ENFERMEDAD NEUROLÓGICA PROGRESIVA CON REGRESIÓN PSICO-MOTORA
SIGNOS Y SÍNTOMAS DE DISFUNCIÓN DE TRONCO O GLANGIOS BASALES: PEO, ataxia, alteraciones respiratorias, nistagmo, distonía, hipotonía y atrofia óptica
AUMENTO DE LACTATO EN SANGRE O LCR
UNO O MÁS DE LOS SIGUIENTES CRITERIOS:
Neuroimagen típica de LS: hipodensidades simétricas en los ganglios basales en TC o lesiones hiperintensas en T2 en RMN
Hallazgos neuropatológicos característicos postmortem
Neuropatología característica en un hermano afectado de forma similar
FACTORES MITOC. RELACIONADOS CON LA CAD. RESP. (NO CLASE)
SPG7: PARAPLEJINA: metaloproteasa mitocondrial: paraplejía espástica
FRATAXINA: proteína de almacenamiento de hierro intramitocondrial; ataxia de Friedreich.
ABC7: exportador de Fe mitocondrial, controlando la generación de proteínas Fe-S citosólicas: Ataxia y anemia sideroblástica ligada al X
DDP1: Componente del sistema de importación de porteínas transportadoras mitocondriales: Síndrome sordera-distonía ligado al X
OPA1: Relacionada con las dinaminas; proteínas que generan vesiculas intracelulares rodeadas de membranas; ad-atrofia óptica y SNPs asociados a glaucoma normotensivo.
T AZ:
homologo de fosfolípido aciltransferasas que controla
metabolismo de cardiolipina, un componente de MIM: Síndrome
de Barth ligado al X
AZ:
homologo de fosfolípido aciltransferasas que controla
metabolismo de cardiolipina, un componente de MIM: Síndrome
de Barth ligado al X
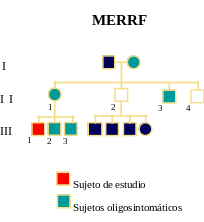
II-1 III-1 III-2 III-3 II-2 Epilepsia
- +++
+ - - Mioclonía
- +++
+ - - Ataxia
- +++
- - - Debilidad
-
-
- - - Neuropatía
periférica +
++
++ ++ - Temblores
- ++
- - - Sordera
- +
- - - Histoquímica
muscular
rrf RRF RRF RRF ND COX-
dispersas dispersas Bioquímica
muscular N Complejo
I
N
N ND
Tags: mitocondrial bq21, encefalomiopatía mitocondrial, patología, mitocondrial, garesse, tiene
- 2 ZAKON O SPREMEMBI IN DOPOLNITVI ZAKONA O USTAVNEM
- MINISTARSTVO MORA TURIZMA PROMETA I RAZVITKA 3059 NA TEMELJU
- REUNIÓN ACLARATORIA DE BASES DE LICITACIÓN MATERIAL DIDÁCTICO Y
- CHECKLIST VOOR UW VERHUIZING HANDIG OM UIT TE PRINTEN!
- US FISH AND WILDLIFE SERVICE [INSERT THE SPECIFIC FUNDING
- SEPTIEMBRE 2018 MESAS DE ASTURIAS 2018 ID CONCEJO (MUNICIPIO)
- ANEXO 1 CONTENIDO DE LA PARTE TEÓRICA IMPARTIDA EN
- CHARGE 510E ― PAGE 2 OF 2 510E
- THE PRESENT DAY GEORGE WASHINGTON CARVER MUSEUM AND CULTURAL
- QUEEN’S GOLDEN JUBILEE CONCERT TICKETS JUNE 2002 72 W
- 8 R E P U B L I K
- IDENTIFICATION OF POSSIBLE SOURCE MARKERS IN MARINE DISSOLVED ORGANIC
- JONAVOS RAJONO SAVIVALDYBĖS ADMINISTRACIJOS DIREKTORIUS ĮSAKYMAS DĖL PASIRENGIMO CIVILINĖS
- BU SCRAPPLE PENN KREME CHEEZE KUP 1997 TOSSUP 1
- 77 MINISTERSTVO ZAHRANIČNÍCH VĚCÍ ČESKÉ REPUBLIKY L
- ADOPTIONS IN GUATEMALA PROTECTION OR BUSINESS? THE CONCENTRATION
- 1 PAGRINDINĖS ŽINIOS APIE MEDŽIAGŲ SUVIRINIMĄ 11 FIZIKINĖ SUVIRINIMO
- H UMAN RIGHTS TRAINING TOOLKIT – 2ND EDITION INTRODUCTION
- XI CONGRÉS CCEPC LA CONSTRUCCIÓ DEL TERRITORI GEOGRAFIA IDENTITAT
- ROMSEY PRIMARY SCHOOL ‘POLICY & PROCEDURES DEVELOPMENT’ POLICY RATIONALE
- KÓRNIK DNIA……………………………… DYREKTOR SZKOŁY PODSTAWOWEJ NR 1 IM TYTUSA
- TINSPIRE™ NAVIGATOR™ VERSIÓN 32 NOTAS DE LA VERSIÓN INTRODUCCIÓN
- LABORATORIEMEDICIN SIDA 2 AV 2 DOKUMENTTYP PROVTAGNINGSINFORMATION DOKUMENTNAMN TOLKNINGSMALL
- 525222-cambridge-pathway-press-release-template
- PROPONOWANE TEMATY BADAŃ STATYSTYCZNYCH PROPOZYCJE Z ROKU AKAD 20062007
- NA OSNOVU ČLANA 22 STATUTA EUROBANK EFG AD BEOGRAD
- ENCUESTA INICIAL HAVE YOU GOT A COMPUTER AT HOME?
- CONCEJO DELIBERANTE DE LA CIUDAD DE CÓRDOBA 21º REUNIÓN
- MISSISSIPPI DEPARTMENT OF EDUCATION OFFICE OF SPECIAL EDUCATION EDUCABLE
- PASANTÍAS EXTERNAS SOLICITUD PARA PRESENTACIÓN DE ANTECEDENTES DATOS
 REGIONAL BIG MEETING IN RAMSAU BERCHTESGADEN MAY 0103
REGIONAL BIG MEETING IN RAMSAU BERCHTESGADEN MAY 0103 ACTA NÚM 21 REUNIÓ DEL 040411 DE LA JUNTA
ACTA NÚM 21 REUNIÓ DEL 040411 DE LA JUNTA27 FUENTES DEL DERECHO Y NORMAS DE ORIGEN JUDICIAL1
 INNEHÅLLSFÖRTECKNING INLEDNING…………………………………………………… SID 1 BESKRIVNING AV UPPDRAGET………………………………… SID 2
INNEHÅLLSFÖRTECKNING INLEDNING…………………………………………………… SID 1 BESKRIVNING AV UPPDRAGET………………………………… SID 2 MINISTERIO DEL INTERIOR E SPAÑA INICIA LA REAPERTURA PROGRESIVA
MINISTERIO DEL INTERIOR E SPAÑA INICIA LA REAPERTURA PROGRESIVA 3 KEMENTERIAN PENDIDIKAN DAN KEBUDAYAAN UNIVERSITAS BRAWIJAYA FAKULTAS ILMU
3 KEMENTERIAN PENDIDIKAN DAN KEBUDAYAAN UNIVERSITAS BRAWIJAYA FAKULTAS ILMU DEVELOPMENTAL DISABILITIES PROGRAM RESIDENTIAL PLACEMENT REFERRAL SUPPORT COORDINATOR
DEVELOPMENTAL DISABILITIES PROGRAM RESIDENTIAL PLACEMENT REFERRAL SUPPORT COORDINATOR GREATER YELLOWSTONE AREA CLEAN AIR PARTNERSHIP ANNUAL MEETING POCATELLO
GREATER YELLOWSTONE AREA CLEAN AIR PARTNERSHIP ANNUAL MEETING POCATELLOPLANTILLA PARA COMENTARIOS SOBRE TRADUCCIONES FECHA DOCUMENTO WCAG 20
ESTABLISHING A BIRTH RECORD CONGRATULATIONS! YOU HAVE JUST GIVEN
 GRUPOS GENERADORES ELÉCTRICOS DE BAJA TENSIÓN GOBIERNO DE CANARIAS
GRUPOS GENERADORES ELÉCTRICOS DE BAJA TENSIÓN GOBIERNO DE CANARIASCONTRATO DE COMPRAVENTA DE VIVIENDA EN …… A ……………DE……………
 DIVISIÓN DE INSTALACIÓN DE CABLE DE FIBRA OPTICA DE
DIVISIÓN DE INSTALACIÓN DE CABLE DE FIBRA OPTICA DEAPPLICATION FOR APPOINTMENT TO A BOARD COMMITTEE OR COMMISSION
 TASK YOU WILL BE ASSIGNED A VOLCANO AND ARE
TASK YOU WILL BE ASSIGNED A VOLCANO AND ARE GRADE 1 SOCIAL STUDIES UNIT 03 LESSON 04 LESSON
GRADE 1 SOCIAL STUDIES UNIT 03 LESSON 04 LESSON HEADLINE BENEFIT NUMBERS AT THE END OF DECEMBER 2009
HEADLINE BENEFIT NUMBERS AT THE END OF DECEMBER 2009 İŞ PLANI DOSYASI (İŞ FİKRİ) İŞ PLANI (GİRİŞİMCİNİN ADI
İŞ PLANI DOSYASI (İŞ FİKRİ) İŞ PLANI (GİRİŞİMCİNİN ADI INEL 4206 SPRING 2002 EXÁMEN II 101921 NOMBRE
INEL 4206 SPRING 2002 EXÁMEN II 101921 NOMBRE  CHUYÊN ĐỀ 5 CHẾ ĐỘ CÔNG VỤ VÀ QUẢN
CHUYÊN ĐỀ 5 CHẾ ĐỘ CÔNG VỤ VÀ QUẢN
